
Dystrofina i jej mutacje - jeden gen, kilka białek i wiele fenotypów - Podstawy molekularne dystrofii mięśniowych Duchenne*a i Becker*a
Anna Dudka | 2005-01-06
Dystrofina jest białkiem kodowanym przez gen leżący na chromosomie X. Opisano wiele mutacji występujących w tym genie, z których większość zmienia ekspresję dystrofiny w komórce. Mutacje te wywołują choroby zwane dystrofiami mięśniowymi Duchenne’a i Becker’a. Jest jeszcze kilka innych tkanek, w których występują izoformy dystrofiny, takie jak tkanka nerwowa czy siatkówka oka. Mutacja jednej z izoform dystrofiny występującej w mięśniach serca powodują kardiomiopatie. Mnogość fenotypów nauczyła naukowców wiele na temat funkcji dystrofiny, jej istotnego znaczenia w komórce, a także niezwykłej roli regulacji ekspresji genów.
Dystrofia mięśniowa Duchenne’a (DMD) jest najczęstszą i najcięższą z postępujących dystrofii mięśniowych (częstość – 1:3600 żywo urodzonych chłopców). Łagodniejszą odmianą jest dystrofia mięśniowa Becker’a (BMD). Dystrofia mięśniowa Duchenne’a była po raz pierwszy opisana przez francuskiego neurologa Guillaume’a Benjamin’a Amand’a Duchenne’a w roku 1860. Dystrofia mięśniowa Becker’a była nazwana przez niemieckiego lekarza Peter’a Emil’a Becker’a w 1950 roku, który po raz pierwszy opisał wariant DMD.
Postępująca dystrofia mięśniowa typu Duchenne’a (DMD) jest chorobą genetyczną powodującą upośledzenie ruchu i bardzo często paraliż kończyn, szczególnie nóg. Po urodzeniu i we wczesnym okresie niemowlęcym, poza niekiedy obserwowaną wiotkością, chorzy chłopcy rozwijają się prawidłowo. Dopiero około drugiego roku życia zmiany genetyczne zaczynają być widoczne. Charakteryzują się one symetrycznym osłabieniem mięśni obręczy biodrowej, kołyszącym, „kaczkowatym” chodem, tendencją do chodzenia na palcach, trudnością we wchodzeniu po schodach i wstawaniu z pozycji leżącej. Odruchy kolanowe początkowo osłabione, z czasem zanikają całkowicie, dłużej utrzymują się odruchy skokowe. Dolegliwości te narastają z wiekiem. Dystrofia mięśniowa Becker’a (DMB) to „bliźniacza” odmiana DMD o niemal jednakowym obrazie klinicznym, lecz o łagodniejszym przebiegu. Jej objawy są widoczne dopiero około 11 roku życia, a nawet dopiero w wieku dojrzałym. Chorzy są długo sprawni, potrafią samodzielnie się poruszać.
W latach 80 odkryto przyczynę obu dystrofii. W roku 1986 naukowcy odkryli, że dystrofie mięśniowe Duchenne’a oraz Beckera powodują mutacje w genie kodującym białko dystrofiny. Gen ten mieści się na chromosomie X, co powoduje, że prawie wyłącznie na tą odmianę dystrofii mięśniowej chorują chłopcy. W przypadku DMD mutacja jest tak duża, iż niemożliwa jest produkcja białka w związku z czym komórki mięśniowe są całkowicie pozbawione dystrofiny w błonie komórkowej. BMD jest powodowana przez inne mutacje w obrębie tego samego genu. Mutacje te umożliwiają produkcję dystrofiny lecz jej jakość i ilość jest również niewystarczająca na prawidłowe funkcjonowanie komórek mięśniowych (jej masa cząsteczkowa jest mniejsza niż dystrofiny prawidłowej). Częściowe zachowanie funkcji dystrofiny może mieć związek z późniejszym wystąpieniem objawów klinicznych oraz z łagodniejszym przebiegiem choroby.
Dystrofia mięśniowa Duchenne’a rozwija się z wiekiem i niezależnie od miejsca wystąpienia mutacji w genie proces ten wygląda tak samo. Rola dystrofiny polega na "uszczelnianiu" błony komórkowej, dzięki czemu zachowuje ona selektywną przepuszczalność (m.in. nie wypuszczając enzymów na zewnątrz, a jonów wapnia do komórki). Dystrofina, którą zawiera mutacje jest niestabilna, co powoduje zaburzenia w utrzymywaniu prawidłowych interakcji pomiędzy dystrofiną a aktyną, przyczyniając się jednocześnie do zaburzeń w strukturze całego cytoszkieletu i powodując zmiany w budowie błony biologicznej. Dochodzi do przechodzenia enzymów (kinaza kreatynowa, adolaza, transaminazy) na zewnątrz błony komórkowej. Powoduje to wzrost ich poziomu, w surowicy, znacznie powyżej normy. Do komórki wnikają natomiast bez przeszkód jony wapnia, co powoduje jej martwicę, stymulację fibroblastów i rozrost tkanki włóknistej w miejscu tkanki mięśniowej. W ten sposób z biegiem czasu coraz więcej włókien jest traconych, prowadzi do degeneracji tkanki mięśniowej. Przez kilka lat tkanka jest zdolna do ciągłej odbudowy włókien, niestety z upływem lat nie jest w stanie dalej się regenerować i włókna mięśniowe zastępowane są tkanką łączną i tłuszczową. Powoduje to stopniowe osłabienie mięśni i problemy z poruszaniem.
Gen kodujący dystrofinę jest największym genem występującym w organizmie ludzkim, zajmuje około 0,1% całego ludzkiego genomu i 1,5% chromosomu X. Składa się w 99% z intronów, natomiast egzonów jest 86. Dystrofina odgrywa istotną rolę w strukturze dużego kompleksu występującego w błonie komórek mięśniowych. Początkowo sądzono, że dystrofina ze względu na swój kształt i rozmiar jest białkiem strukturalnym, zapewniającym wytrzymałość błony komórkowej mięśni. Dokładniejsze badania ustaliły, iż jest to tylko jedna z funkcji tego białka, dystrofina chroni również komórki mięśniowe przed nekrozą, a także pełni rolę pośrednika podczas komunikacji międzykomórkowej (jest fosforylowana przez kinazy prolinowe, serynowo-treoninowe i kalmodulinozależne) oraz „uszczelnia” błonę biologiczną. Dystrofina występuje w postaci izoform, co związane jest z miejscem występowania. Ekspresja genu kodującego dystrofinę zachodzi w kilku tkankach, min w mięśniach gładkich, szkieletowych i sercowych, a także w siatkówce, mózgu i neuronach. Izoformy długie dystrofiny występują w tkance mięśniowej, natomiast krótki izoformy dystrofiny w pozostałych tkankach.
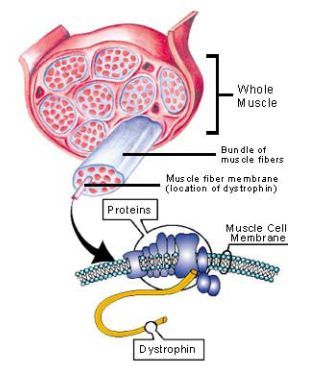
Rys.1 Lokalizacja dystrofiny w tkance mięśniowej. (http://www.mdausa.org/publications/fa-dmdbmd-what.html)
Dystrofina zbudowana jest z 3685 aminokwasów o wadze 427 kDa. Podzielona jest na cztery domeny - pierwsza domena na N-końcu o wadze 30 kDa jest homologiem α-aktyniny, domena centralna zbudowana z 25 powtórzonych motywów potrójnych helis podobnych do występujących w spektrynie, domena bogata w cysteiny (280 reszt) oraz C-końcowa domena. C-końcowa domena zawiera kilka charakterystycznych motywów – domenę WW (posiadają dwie konserwatywne reszty tryptofanu), motyw EF hands, domenę ZZ (motyw wiążący cynk) oraz dwie α-helisy. Krótkie izoformy są pozbawione domeny N-końcowej, wiążącej F-aktynę, natomiast zostaje domena bogata w cysteiny i domena C-końcowa, wiążąca dystroglikany, dystrobrewinę i syntrofinę. Dystrofina stabilizuje sarkomer łącząc się ze szkieletem aktynowym oraz kompleksem glikoproteinowym po stronie zewnętrznej komórki. Jej N-koniec wiąże F-aktynę, natomiast C-koniec wiąże kompleks glikoproteinowy DAG zlokalizowany w błonie. Kompleks glikoproteinowy składa się z mniejszych kompleksów dystroglikanów i sarcoglikanów. Dystroglikany łączą dystrofinę z lamininą-α2. Sarkoglikany są zbudowane z czterech równomolarnych, transbłonowych glikoprotein o nieznanej funkcji. Dystrofina tworzy swoiste połączenie pomiędzy sarkoplazmatycznym cytoszkieletem a zewnątrzkomórkowym matrix przyczyniając się do utrzymania homeostazy w komórce.
Na N-końcu dystrofiny znajduje się region bardzo podobny do jednej z domen białek rodziny spektryn, które wiążą się z F-aktyną, do tej grupy białek należą β-spektryna, α-aktynina, filamina i ABP120. Badania ujawniły, że również natywna dystrofina wiąże F-aktynę in vitro. Także białka fuzyjne zawierające część aminokwasów z N-końca dystrofiny (co najmniej 233 aminokwasy) są w stanie wiązać aktynę. Spekuluje się, iż trzy regiony w obrębie dystrofiny są odpowiedzialne za wiązanie aktyny. Są to miejsca określone jako ABS1, ABS2 i ABS3. ABS1 i ABS2 są przypuszczalnie miejscami interakcji pomiędzy dystrofiną a aktyną, natomiast ABS3 jest wysoce konserwatywną sekwencją.
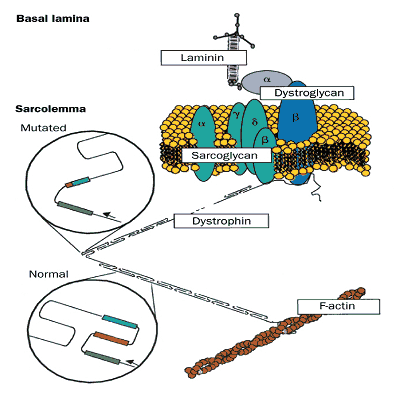
Rys.2 Molekularna organizacja dystrofiny w połączeniu z kompleksem glikoproteinowym oraz F-aktyną. Pokazana jest normalna i zmutowana struktura dystrofiny (Association of nonsense mutation of dystrophin gene with disruption of sarcoglycan complex in X-linked dilated cardiomyopathy, Franz W.M. et al Lancet 2000; 355: 1781-85).
Aby zdecydować, czy wszystkie trzy sekwencje są istotne dla wiązania aktyny przeprowadzono szereg doświadczeń, w których analizowano cztery N-końcowe białka fuzyjne dystrofiny, do których wprowadzano odpowiednie mutacje eliminujące miejsca wiązania aktyny. Wyniki sugerują, że ABS2 i ABS3 nie są niezbędne do wiązania aktyny przez dystrofinę. Sekwencja ABS1 wykazywała największą zdolność do wiązania aktyny lecz oprócz tych trzech sekwencji badania ujawniły inną sekwencję - czwartą, prawdopodobnie najistotniejszą.
Znając miejsca wiązania aktyny przez dystrofinę można badać molekularne podstawy dystrofii Duchenne’a i Becker’a. Rearanżacja genów, która występuje u około ¾ pacjentów, jest wynikiem dużych delecji i duplikacji oraz mutacji punktowych. Delecje są bardzo pospolitymi zmianami w genie dystrofiny. Stanowią około 65% ogólnych mutacji. Duplikacje występują nieco rzadziej (5-15% pacjentów). Mutacje punktowe pojawiają się u około 20-35% pacjentów. Metody detekcji mutacji występujących w genie dystrofiny powodujących choroby genetyczne wykazały największą ilość zmian w N-końcu dystrofiny. Koniec ten jest odpowiedzialny za wiązanie aktyny. Dystrybucja mutacji – dużych delecji i duplikacji nie jest więc przypadkowa.
Istnieje wiele przykładów delecji części genu. Najczęściej mutowany region to domena N-końcowa, a dokładnie egzony 2-19 oraz dalej położone egzony 45-55. Delecja egzonów 2-7, 3 i często 3-7 objawia się przeważnie u chorych na BMD. Delecja w tych egzonach prowadzi do skrócenia dystrofiny, której brakuje od 11 do 217 aminokwasów znajdujących się w ABS1-3.
Nie można jednoznacznie ustalić zależności pomiędzy rodzajem i miejscem mutacji a zmianą fenotypu. Produkt jednego genu może budować kilka fenotypów. Niezwykle istotna jest tu regulacja ekspresji genu. W tym przypadku jest to proces bardzo skomplikowany i nie do końca zrozumiany. Przykładem zależności między genotypem a fenotypem jest delecja małego fragmentu egzonu 44 powodująca DMD, natomiast duża delecja, która zajmuje ponad połowę całego genu, powoduje łagodniejszą chorobę – BMD. Pacjenci z delecją egzonów – 32-44, 48-51 lub 48-53 wszyscy mają normalny lub prawie normalny poziom dystrofiny. Efekt mutacji zależy więc nie od ilości delecji, ale od tego czy delecja ta zaburza ramkę odczytu czy też nie. Kolejną obserwacją jest również to, że różne mutacje, w różnych miejscach mogą dawać bardzo podobne fenotypy.
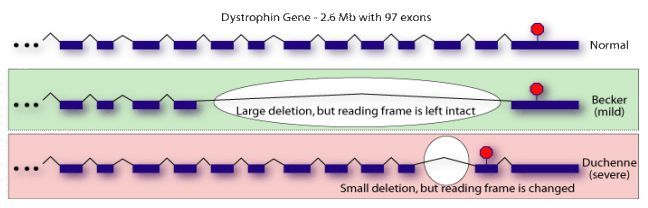
Rys. 3 Porównanie delecji wywołującej dystrofie Becker’a i Duchenne’a (http://compbio.berkeley.edu/people/ed/rust/Dystrophin.html).
Zjawisko to jest tłumaczone za pomocą hipotezy zmiany ramki odczytu (frame-shift hypotesis). Mutacje, które pozwalają zachować ramkę odczytu (tzw. mutacje in-frame) dają produkt w postaci zmienionej dystrofiny lecz nadal prawidłowo funkcjonującej i przeważnie mutacja tego typu wywołują dystrofię Becker’a. U pacjentów z DMD mutacja zaburza ramkę odczytu (tzw. mutacja frame-shift), co doprowadza do produkcji niestabilnego RNA, które daje w rezultacie bardzo niskie stężenie skróconego białka. Ta hipoteza potwierdza większość przypadków DMD i BMD (około 90% przypadków).
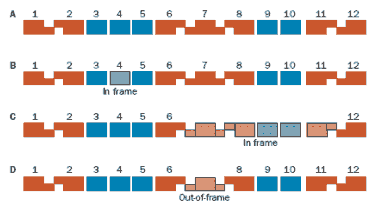
Rys. 4 Wpływ różnych delecji na ramkę odczytu genu dystrofiny (A). Delecja egzonu 4 (B), egzonów 7-11 (C) zachowuje ramkę odczytu. Delecja egzonu 7 prowadzi do utraty ramki odczytu (D) (Dystrophin and mutations: one gene, several proteins, multiple phenotypes, Muntoni F, Torelli S, and Ferlini A, Lancet Neurol 2003; 2: 731–40).
Oczywiście występują wyjątki, są także pacjenci, którzy mimo mutacji naruszającej ramkę odczytu chorują na BMD oraz pacjenci, którzy chorują na DMD pomimo mutacji typu in-frame. Mutacje w egzonach 5-7 lub 3-6 znajdujących się na N-końcu lub w egzonach dalej położonych – 51, 49-50, 47-52, 44 lub 45 prawie zawsze powodują DMD (przesunięcie ramki odczytu). Czasem jednak pacjenci z takimi mutacjami przechodzą łagodniejszą odmianę dystrofii czyli BMD. Dzieje się tak dzięki procesowi zwanemu „przeskakiwaniem egzonu” (exon skipping) - alternatywny splicing. Polega to na pominięciu miejsca delecji egzonu i prowadzeniu dalszej transkrypcji. Jest to możliwe dzięki dodatkowemu miejscu startu transkrypcji. Podstawy tego mechanizmu nie są jednak znane i jest on nadal niejasny dla naukowców.
Zdarzają się także mutacje, które nie zaburzają ramki odczytu (mutacje in-frame) lecz mimo to powodują DMD. Jest to w większości związane z mutacją w egzonie 3-13, gdzie kodowana jest N-końcowa część dystrofiny, wiążąca się z aktyną.
Przykładem mutacji punktowych są pacjenci z objawami DMD, którzy zamiast Leu54 posiadają Arg. Mutacja ta nie jest zbyt obszerna lecz zmienia ona ramkę odczytu, co prowadzi do zmiany w budowie dystrofiny. Przeważnie większość chorych produkuje dystrofinę w bardzo małych ilościach lub w ogóle. Mutacje punktowe pojawiające się u pacjentów z BMD to najczęściej Ala168-Asp i Tyr231-Asn oraz Ala171-Pro. Mutacje punktowe są rozłożone bardziej równomiernie w genie, część z nich również znajduje się na N-końcu.
Dystrofina jest niezwykle istotnym komponentem błony biologicznej. Dość charakterystyczne jest to, iż z jednego genu może powstać kilka izoform białka. Gen kodujący dystrofinę jest ogromnym i fascynującym genem charakteryzującym się złożonością regulacji transkrypcji, funkcji i oddziaływań białko-białko. Dopiero powoli zaczynamy rozumieć zależności i mechanizmy funkcjonowania genu i jego produktu – dystrofiny. Relacja pomiędzy genotypem a fenotypem jest ważna nie tylko ze względu na diagnostykę lecz także ze względu na zrozumienie mechanizmu regulacji ekspresji genów. Udoskonalanie wiedzy w tym zakresie pozwoli na stworzenie w przyszłości metody leczenia dystrofii mięśniowych.
Literatura:
1. Dystrophin and mutations: one gene, several proteins, multiple phenotypes, Muntoni F, Torelli S, and Ferlini A, Lancet Neurol 2003; 2: 731–40
2. The structure of the N-terminal actin-binding domain of human dystrophin and how mutations in this domain may cause Duchenne or Becker muscular dystrophy, Fiona LM Norwood F.L. et al Elsevier 2000, 8:481–491
3. Association of nonsense mutation of dystrophin gene with disruption of sarcoglycan complex in X-linked dilated cardiomyopathy, Franz W.M. et al Lancet 2000; 355: 1781-85
4. Deletion analysis of the dystrophin-actin binding domain, Corrado K. et al FEBS Letters 344 (1994) 255-260
5. http://www.ygyh.org/dmd/cause.htm
6. http://compbio.berkeley.edu/people/ed/rust/Dystrophin.html
7. http://www.hprd.org/protein/02303?selectedtab=INTERACTIONS
8. http://www.mda.org.au/specific/mdadmd.html
Słowa kluczowe: choroby genetyczne, wrodzone, dystrofina, dystrofia mięśniowa, DMD, BMD